Kompiuterinė chemija: apibrėžimas, metodai ir praktiniai taikymai
Kompiuterinė chemija: apibrėžimas, metodai ir praktiniai taikymai — kaip skaičiavimai kuria naujus vaistus, pažangias medžiagas ir prognozuoja molekulių struktūras bei savybes.
Kompiuterinė chemija - tai chemijos šaka, kurioje cheminės problemos sprendžiamos pasitelkiant kompiuterių mokslą. Naudojamos programos apskaičiuoja molekulių ir kietųjų kūnų struktūras, energetiką ir kitas savybes. Kompiuterinė chemija dažnai papildo ir interpretuoja cheminius eksperimentus, leidžia numatyti cheminius reiškinius ir yra plačiai taikoma kuriant naujus vaistus bei medžiagas.
Vaizdų galerija
8 Vaizdai


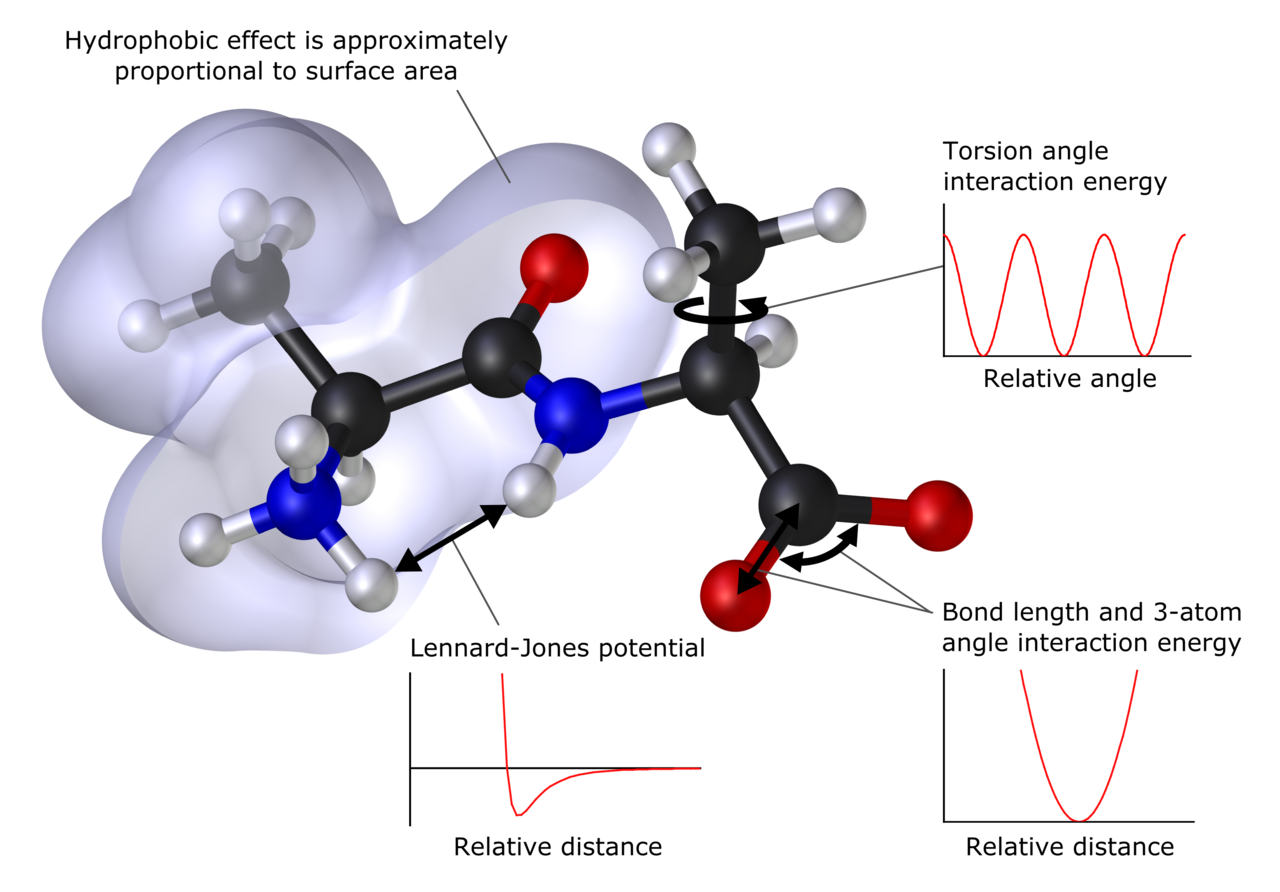


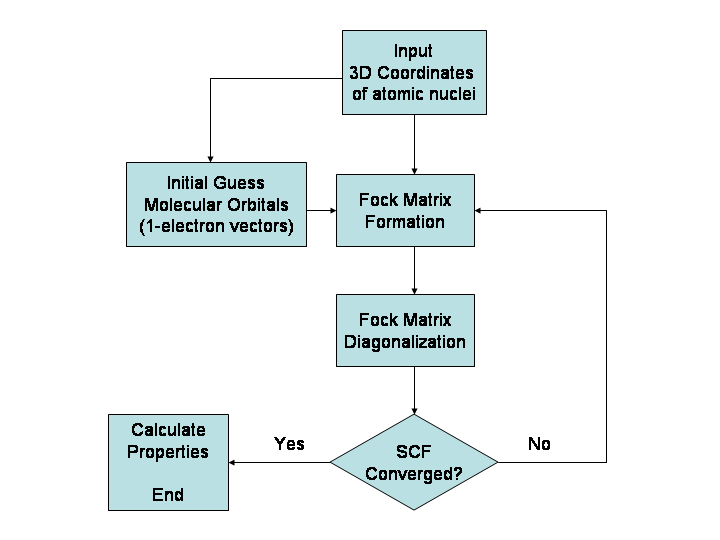
Apibrėžimas ir ką galima apskaičiuoti
Kompiuterinė chemija gali numatyti molekulių ir kietųjų kūnų atomų padėtį (struktūrą), absoliučią ir santykinę (sąveikos) energiją, elektroninių krūvių pasiskirstymą, dipolius ir aukštesniuosius daugiapolius momentus, virpesių dažnius, reaktyvumą bei kitus spektroskopinius dydžius. Taip pat galima skaičiuoti susidūrimo su kitomis dalelėmis skerspjūvius, kinetiką, termodinamines savybes ir elektroninio perėjimo procesus.
Pagrindiniai metodai
Kompiuterinės chemijos metodai skirstomi pagal tikslumą ir skaičiavimo sąnaudas. Dažniausiai naudojami:
- Ab initio kvantiniai metodai (pvz., Hartree–Fock, MP2, CCSD) – labai tiksli teorija, bet sparčiai didėja skaičiavimo kaina su sistemos dydžiu.
- Funkcionalų teorija (DFT) – geras kompromisas tarp tikslumo ir išteklių; plačiai naudojama elektroninei struktūrai ir reaktyvumui nagrinėti.
- Pusiauempiriniai metodai – supaprastinti kvantiniai metodai, tinkami didesnėms molekulėms ir greitoms apžvalgoms.
- Molekulinė mechanika (MM) – klasikiniai modeliai, kur energija aprašoma jėgų laukais; leidžia nagrinėti dideles sistemas ir ilgas laiko skalės dinamikas.
- Molekulinė dinamika (MD) – laiko evoliucijos simuliacijos naudojant klasikinę ar kvantinę mechaniką (QM/MM hibridai), tinka procesams, priklausantiems nuo laiko, pvz., baltymų judėjimui.
- Monte Carlo metodai – statistiniai metodai konfigūracijų rinkiniams ir termodinaminėms savybėms apskaičiuoti.
Taikymai
Praktiniai kompiuterinės chemijos taikymai yra labai įvairūs:
- Vaistų atranka ir ligando optimizavimas (ligandų pakabinimo modeliavimas, farmakoforų paieška).
- Medžiagų projektavimas (pvz., puslaidininkiai, katalizatoriai, polimerai, baterijų medžiagos).
- Spektroskopijos interpretacija (IR, Raman, NMR, UV–Vis, elektroninės spektroskopijos apskaičiavimai).
- Reakcijų mechanizmų ir energijos barjerų nustatymas bei cheminių kinetikų modeliavimas.
- Fazinių perėjimų ir kietųjų medžiagų savybių tyrimai.
Praktinė darbo eiga
Tipinė kompiuterinio tyrimo eiga:
- Modelio kūrimas: cheminės struktūros sudarymas arba duomenų importas.
- Geometrijos optimizavimas: randama energiją mažiausią konfigūraciją.
- Frekvencijų skaičiavimai: patikrinama, ar rasta struktūra yra stabilus minimumas (ne turi nerealių virpesių) arba pereinamasis taškas.
- Elektroninės struktūros analizė: orbitalės, krūviai, energijų skirtumai.
- Ekstra: solvatacijos modeliai, reagento koncentracijos ir temperatūros įtaka, termodinaminiai skaičiavimai.
- Rezultatų vertinimas ir validacija su eksperimentiniais duomenimis.
Ribotumai ir klaidų šaltiniai
Kompiuterinė chemija yra galinga, bet turi apribojimų:
- Skaliarinė priklausomybė: daug tikslumo reikalaujančių metodų taikymas didelėms sistemoms yra praktiškai neįmanomas dėl laiko ir atminties reikalavimų.
- Aproksimacijos: kiekvienas metodas turi prielaidų (pvz., DFT funkcionalų netobulumas, baigtinių bazės klaidos, jėgų laukų parametrizacija).
- Modelio parinkimas: neteisingas modelis ar užuomazga gali duoti klaidingas išvadas.
- Solvatacija ir temperatūros efektai: jų neįvertinimas gali stipriai pakreipti rezultatus.
Programinė įranga ir ištekliai
Yra daugybė programų ir bibliotekų kompiuterinei chemijai: tiek komercinių, tiek atviro kodo sprendimų. Populiariausi pavyzdžiai apima kvantinių skaičiavimų programas (pvz., Gaussian, ORCA, GAMESS, NWChem, VASP) ir molekulinių dinaminių paketų (pvz., GROMACS, LAMMPS). Darbo našumui gerinti naudojamos GPU akceleracijos, aukštos klasės skaičiavimo sistemos ir debesų resursai.
Patarimai pradedantiesiems
- Pradėkite nuo paprastų sistemų ir palyginkite įvairius metodus, kad suprastumėte jų skirtumus.
- Visada tikrinkite optimizuotų struktūrų vibracijas — jos padeda atskirti minimumus nuo pereinamųjų taškų.
- Validuokite skaitinius rezultatus su eksperimentiniais duomenimis, kai tik tai įmanoma.
- Domėkitės literatūra ir bendruomenės gairėmis dėl tam tikrų metodų taikymo skirtingoms sistemoms.
Ateities kryptys
Kompiuterinė chemija sparčiai vystosi: daugėja hibridinių QM/MM metodų, mašininio mokymosi modelių, kurie greitai prognozuoja savybes, ir didelio masto multiskalinių simuliacijų. Taip pat tobulėja funkcionalai, jėgų laukai ir skaičiavimo aparatūra (pvz., kvantiniai skaičiuotuvai ateityje gali atverti naujas galimybes).
Apibendrinant, kompiuterinė chemija suteikia plačias galimybes suprasti ir prognozuoti chemines sistemas, tačiau reikalingas atsargumas renkantis metodus ir nuolatinė rezultatų validacija su eksperimentais.

Susiję puslapiai
- Bioinformatika
- Statistinė mechanika
Klausimai ir atsakymai
K: Kas yra skaičiavimo chemija?
A: Skaičiavimo chemija - tai chemijos šaka, kurioje cheminėms problemoms spręsti naudojama kompiuterija. Ji gali būti naudojama molekulių ir kietųjų kūnų struktūroms ir savybėms apskaičiuoti, dar nepastebėtiems cheminiams reiškiniams prognozuoti, naujiems vaistams ir medžiagoms kurti.
K: Kokių tipų sistemas nagrinėja kompiuterinė chemija?
A.: Kompiuterinė chemija nagrinėja ir statines, ir dinamines sistemas. Sistema gali būti atskira molekulė, molekulių grupė arba kieta medžiaga.
K: Kokio pobūdžio informaciją gali suteikti kompiuterinė chemija?
A: Kompiuterinė chemija gali suteikti tokią informaciją kaip struktūra (atomų padėtis), absoliučios ir santykinės energijos, elektroninių krūvių pasiskirstymas, dipoliai ir aukštesnieji daugiapoliai momentai, virpesių dažniai, reaktyvumas ar kiti spektroskopiniai dydžiai ir susidūrimo su kitomis dalelėmis skerspjūviai.
Klausimas: Kiek tikslūs yra skaičiavimo chemijoje naudojami metodai?
A: Skaičiuojamojoje chemijoje taikomų metodų tikslumas svyruoja nuo labai tikslių iki labai apytikslių. Labai tikslūs metodai paprastai taikomi tik mažoms sistemoms.
K: Kaip kompiuterinė chemija papildo eksperimentinius duomenis?
A.: Kompiuterinė chemija paprastai papildo cheminiais eksperimentais gautą informaciją. Ji gali būti naudojama siekiant numatyti rezultatus, kurie dar nebuvo pastebėti eksperimentiškai.
K: Ar tiriamos sistemos dydis turi įtakos tam, kiek reikia kompiuterio darbo laiko?
A: Taip - didėjant tiriamos sistemos dydžiui, didėja ir analizei atlikti reikalingo kompiuterio darbo laiko sąnaudos, taip pat didėja tokie ištekliai, kaip atmintis ir disko vieta, reikalinga saugojimui.
Susiję straipsniai
Autorius
AlegsaOnline.com Kompiuterinė chemija: apibrėžimas, metodai ir praktiniai taikymai Leandro Alegsa
URL: https://lt.alegsaonline.com/art/22297