Talasemija: genetinis anemijos sutrikimas — simptomai ir gydymas
Talasemija: sužinokite apie šio genetinio anemijos sutrikimo simptomus, diagnostiką, gydymo galimybes, komplikacijas ir prevenciją.
Talasemija (arba talasemija) yra genetinis kraujo sutrikimas, kilęs iš Viduržemio jūros regiono.
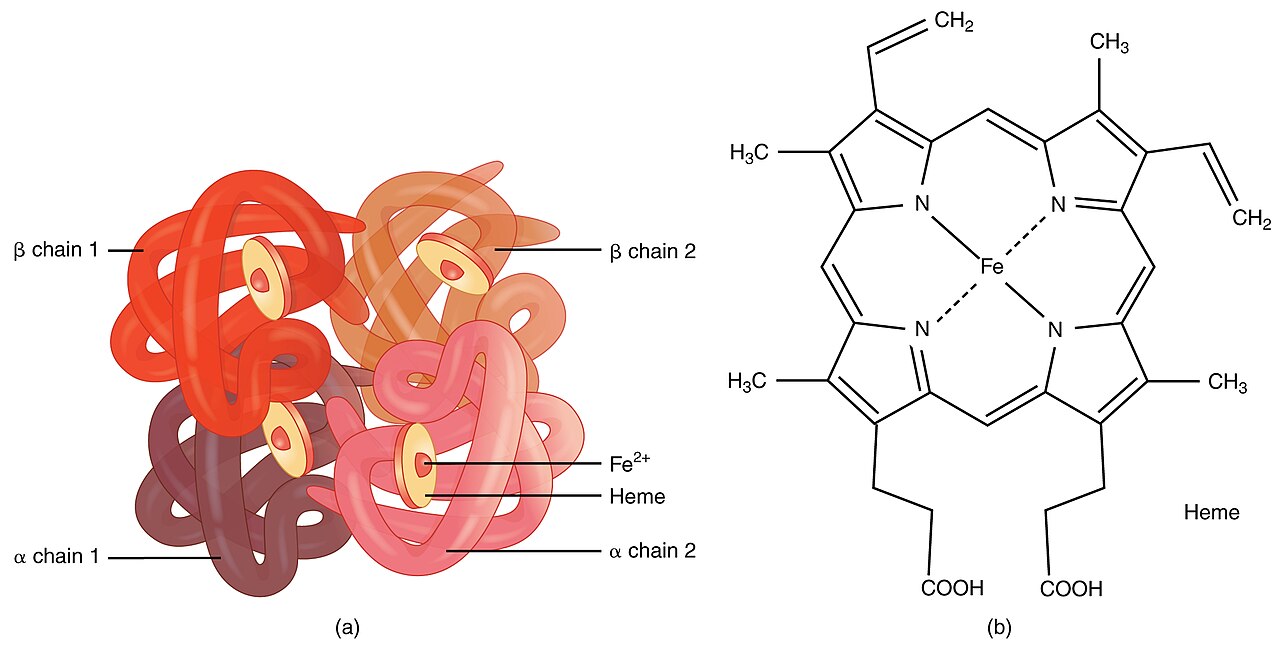
Šią ligą sukelia raudonųjų kraujo kūnelių susilpnėjimas ir sunaikinimas. Tai sukelia mutavę genai, kurie turi įtakos tam, kaip organizmas gamina hemoglobiną. Hemoglobinas yra raudonųjų kraujo kūnelių baltymas, pernešantis deguonį. Sergantieji talasemija gamina mažiau hemoglobino ir mažiau cirkuliuojančių raudonųjų kraujo kūnelių nei įprastai, todėl susergama lengva arba sunkia anemija.
Talasemija gali sukelti rimtų komplikacijų, įskaitant plaučių uždegimą, geležies perkrovą, kaulų deformacijas ir širdies ir kraujagyslių ligas. Tačiau ši paveldima raudonųjų kraujo kūnelių liga suteikia tam tikrą apsaugą nuo maliarijos, kuri yra arba buvo paplitusi tuose regionuose, kur šis požymis yra paplitęs. Šis atrankinis išlikimo pranašumas nešiotojams (vadinamas heterozigotiniu pranašumu) lemia, kad mutacija populiacijose išlieka gerokai didesnė už jos mutacijų dažnį. Talasemijos alelio nešiotojai yra heterozigotai, t. y. tik vienas iš dviejų jų alelių yra mutavęs (nenormalus). Yra keletas skirtingų talasemijos versijų. Kiekvieną jų sukelia mutacija skirtingoje genomo vietoje.
Šiuo požiūriu talasemija panaši į kitą genetinį sutrikimą, susijusį su hemoglobinu, t. y. į serpantininių ląstelių ligą.
Talasemija sergančius ligonius galima išgydyti persodinus suderinamų donorų kaulų čiulpus. Tačiau šiam metodui reikia HLA suderinamo donoro.
Vaizdų galerija
8 Vaizdai





Kas sukelia talasemiją?
Talasemiją sukelia mutacijos genuose, atsakinguose už hemoglobino alfa ar beta grandinių sintezę. Pagal paveiktą globino grandinę skiriama:
- alfa-talasemija – pažeista alfa grandinė;
- beta-talasemija – pažeista beta grandinė.
Mutacijos paveldimos autosominio recesyvinio tipo būdu: kad pasireikštų sunki forma, paprastai reikia, kad abu tėvų perdavimo aleliai būtų pakitę. Asmenys su vienu pakitusiu aleliu yra nešiotojai (heterozigotai) ir dažnai serga lengvesne forma arba yra be simptomų.
Kur talasemija paplitusi?
Talasemija dažnesnė Viduržemio jūros regione, Artimuosiuose Rytuose, Pietų Azijoje, Pietryčių Azijoje ir kai kuriose Afrikos dalyse. Dėl migracijos atvejų talasemijos nešiotojų ir sergančiųjų galima sutikti visame pasaulyje.
Simptomai
Simptomų spektras labai įvairuoja, priklausomai nuo ligos tipo ir sunkumo:
- lengva anemija arba visiškas jos nebuvimas (nešiotojai);
- nuovargis, blyškumas, silpnumas;
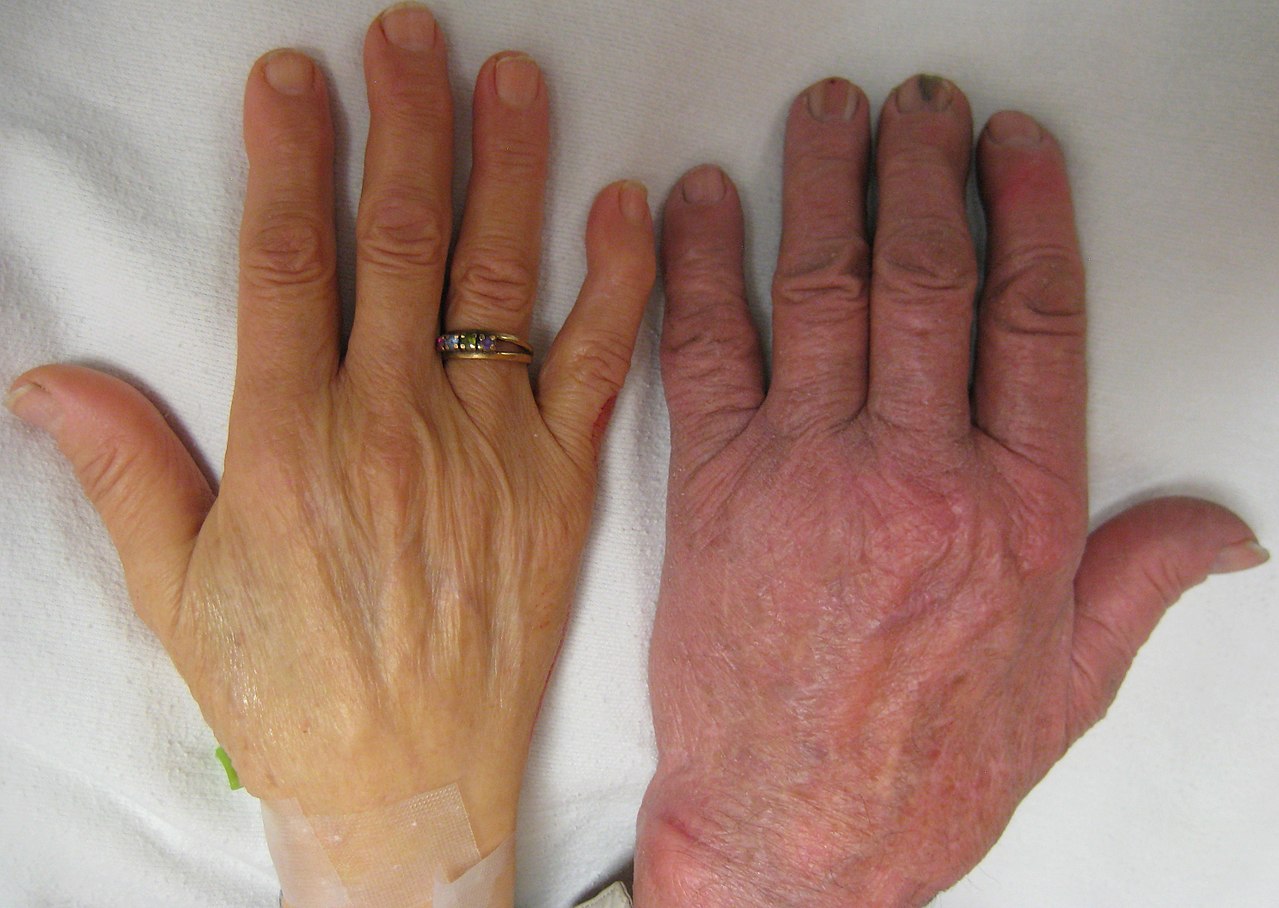
- gelta (odos ir akių pageltimas) dėl padidėjusio hemolizės;
- veiksnio sulėtėjimas vaikams, augimo atsilikimas;
- padidėjęs kepenų ir blužnies tūris (hepatosplenomegalija);
- kaulų deformacijos (ypač kaukolės ir veido kaulų iškraipymas) dėl padidėjusio kaulų čiulpų aktyvumo;
- dažnos infekcijos dėl imuninės sistemos pažeidimų ar po savo blužnies pašalinimo;
- geležies perteklius (hemosiderozė) – dažna gydymo komplikacija po ilgalaikių kraujo perpylimų.
Diagnozė
Diagnozei nustatyti naudojami tam tikri tyrimai:
- bendras kraujo tyrimas (CBC) – dažnai matyti mikrociutinė, hipochrominė anemija, sumažėjęs hemoglobinas, mažas vidutinis korpuskulinis tūris (MCV);
- periferinio kraujo tepinėlis – būdingi „taško“ (target) eritrocitai, anisocitozė, poikilocitozė;
- hemoglobino elektroforezė arba HPLC – nustato hemoglobino izoformas (pvz., padidėjęs HbA2 ir/ar HbF beta-talasemijoje);
- geležies parametrai (feritinas) – padeda įvertinti, ar yra geležies perteklius;
- molekuliniai genetiniai tyrimai – identifikuoja konkrečias mutacijas ir patvirtina tipą.
Gydymas
Gydymo strategija priklauso nuo talasemijos tipo ir sunkumo:
- lengvos formos dažnai nereikalauja specifinio gydymo – stebima būklė, rekomenduojama folio rūgštis;
- vidutinio sunkumo – gali prireikti retų transfuzijų ir nuolatinio stebėjimo;
- sunkios formos (pvz., Cooley anemija, beta-talasemijos major) – būtinos reguliarios raudonųjų kraujo kūnelių perpylos, kad palaikyti normalią hemoglobino koncentraciją;
- geležies kelio terapija (cheliacijos gydymas) po dažnų perpylimų – vaistai kaip deferoksaminas, deferasiroksas ar deferipronas padeda šalinti perteklinę geležį;
- splenektomija kartais atliekama pacientams su dideliu blužnies veikimu ir dažnomis transfuzijų poreikiais;
- kaulų čiulpų / hematopoetinių kamieninių ląstelių transplantacija – vienintelis dabar plačiai prieinamas gydymo būdas, galintis visiškai išgydyti talasemiją, tačiau reikalauja suderinamo HLA donoro ir turi rizikų;
- genų terapija – intensyviai tiriama; pastaraisiais metais pasiekta pažanga, tačiau ši terapija dar nėra universaliai prieinama;
- profilaktinės priemonės – vakcinacija, infekcijų gydymas, mitybos stebėjimas ir periodinė specialisto priežiūra.
Komplikacijos
Be anemijos, svarbios komplikacijos yra:
- geležies perteklius – gali pažeisti širdį, kepenis, endokrininę sistemą (pvz., cukrinis diabetas, hipogonadizmas);
- širdies nepakankamumas ir kitos širdies ir kraujagyslių problemos;
- kaulų deformacijos ir augimo sutrikimai;
- padidėjusi jautrumas infekcijoms;
- psichosocialiniai sunkumai dėl lėtinės ligos ir intensyvios terapijos poreikio.
Prevencija ir genetinis konsultavimas
Dėl paveldimumo svarbu atlikti nešiotojų patikrinimus ir genetinį konsultavimą poroms, kurios planuoja vaiką. Galimos priemonės:
- nešiotojų tyrimai (krūvinės šeimos istorijos ir laboriniai tyrimai);
- prenatalinė diagnostika – choriono ląstelių tyrimas arba amniocentezė genetinėms mutacijoms aptikti;
- paramos programos ir informacija apie galimas reprodukcines pasirinktis (pvz., preimplantacinė genetinė diagnostika IVF metu).
Kasdienė priežiūra ir stebėsena
Žmonėms, sergantiems talasemija, rekomenduojama:
- reguliariai lankytis pas hematologą;
- stebėti geležies rodiklius (feritinas, kepenų funkcija, kartais širdies T2* MR skenavimas);
- nešiojant gydymo kortelę ar informuojant apie ligą sveikatos priežiūros įstaigoms, ypač prieš procedūras ar operacijas;
- vengti savarankiško geležies papildų vartojimo be gydytojo nurodymo (jei nėra įrodytos geležies stokos);
- skiepytis pagal rekomendacijas (ypač prieš spleenektomiją) ir laiku gydyti infekcijas.
Prognozė
Prognozė priklauso nuo talasemijos tipo ir gydymo prieinamumo. Su modernia transfuzine terapija, geležies cheliacija ir specializuota priežiūra daugelis pacientų gyvena iki pilnametystės ir toliau turi geresnę gyvenimo kokybę nei anksčiau. Tačiau sunkios formos be tinkamo gydymo liks gyvybei pavojingos.
Jeigu įtariate, kad esate talasemijos nešiotojas arba liga pasireiškė šeimoje, kreipkitės į šeimos gydytoją arba hematologą dėl reikalingų tyrimų ir genetinio konsultavimo.
Klausimai ir atsakymai
K: Kas yra talasemija?
A: Talasemija yra genetinis kraujo sutrikimas, atsiradęs Viduržemio jūros regione. Šią ligą sukelia raudonųjų kraujo kūnelių susilpnėjimas ir sunaikinimas dėl mutavusių genų, kurie turi įtakos tam, kaip organizmas gamina hemoglobiną.
K: Kokios yra su talasemija susijusios komplikacijos?
A: Su talasemija susijusios komplikacijos gali būti plaučių uždegimas, geležies perkrova, kaulų deformacijos ir širdies ir kraujagyslių ligos.
K: Kaip talasemija apsaugo nuo maliarijos?
A: Talasemijos nešiotojai turi selektyvų išgyvenimo pranašumą nešiotojams (vadinamąjį heterozigotinį pranašumą), kuris padeda išlaikyti šią mutaciją populiacijose gerokai viršijančią jos mutacijų dažnį. Tai suteikia jiems apsaugą nuo maliarijos, kuri yra arba buvo paplitusi tuose regionuose, kur šis požymis yra paplitęs.
Klausimas: Ar yra skirtingų talasemijos versijų?
A: Taip, yra keletas skirtingų talasemijos versijų, kurių kiekvieną sukelia mutacija skirtingoje genomo vietoje. Ji panaši į kitą genetinį sutrikimą, susijusį su hemoglobinu, t. y. į serpantininių ląstelių ligą.
Klausimas: Ar įmanoma išgydyti talasemija sergančius pacientus?
Atsakymas: Taip, talasemija sergančius pacientus galima išgydyti persodinant kaulų čiulpus iš suderinamų donorų, kurie turi HLA suderinamą donorą.
Susiję straipsniai
Autorius
AlegsaOnline.com Talasemija: genetinis anemijos sutrikimas — simptomai ir gydymas Leandro Alegsa
URL: https://lt.alegsaonline.com/art/97356
Šaltiniai
- accessmedicine.com : accessmedicine.com/content.aspx?aID=6123722
- mayoclinic.com : mayoclinic.com/health/thalassemia/DS00905/DSECTION=complications
- bloodjournal.hematologylibrary.org : HLA-matched sibling bone marrow transplantation for β-thalassemia major